台灣出生12日男嬰確診患 罕見遺傳病 獲全額資助$1200萬買藥治療
2023-10-18

罕見遺傳病 |脊髓肌肉萎縮症(SMA)是一類遺傳性神經肌肉疾病,受影響的病童會在運動、飲食、呼吸方面逐漸出現困難。SMA的病發率約為10萬分之1,最常見及非常嚴重的一型,病徵於6個月大之前已經非常明顯,最終因呼吸衰竭在2歲前死亡,是目前嬰兒死亡率最高的遺傳疾病。
台灣一名男嬰於出生12日時確診SMA,雖然可以注射一次性基因治療藥物以免於死亡威脅或運動功能退化,但1針費用高達4,900萬元新台幣(約1,200萬港元),因而令其父母感到徬徨;幸好當地於今年8 月將藥物納入全民健康保險(健保),讓男嬰成為於台灣接受資助的首例,目前治療效果良好。
台灣出生12日男嬰確診患 罕見遺傳病 獲全額資助$1200萬買藥治療

-
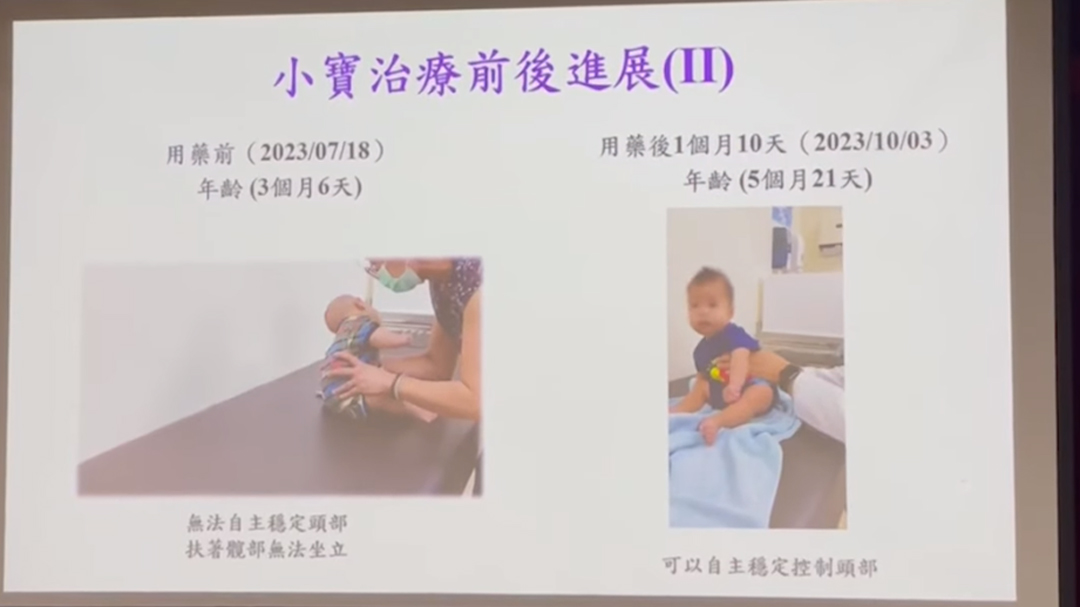
- 台灣一名男嬰於出生12日時確診SMA,用藥前(左)頭部無法直立,用藥後1個月左右(右),已能可以抬頭。(影片截圖)
-

- 男嬰用藥前(左)全身軟弱無力,用藥後1個月左右(右),已能可以連續翻身、自己抬頭。(影片截圖)
-

- 男嬰可以注射一次性基因治療藥物以免於死亡威脅或運動功能退化,但1針費用高達4,900萬元新台幣(約1,200萬港元),幸好當地將藥物納入資助項目,對此男嬰父親表示非常感謝。(影片截圖)
-

- 高雄醫學大學附設中和紀念醫院教授鐘育志指,SMA曾經被視為無法醫治的神經退化疾病,現時則有藥物治療,不過由於一次性基因治療藥物費用高昂,因此男嬰接受注射的過程中,醫療團隊都小心翼翼地進行。(影片截圖)
-

- 台灣健保署指,為有效預防及治療SMA,新生嬰兒應及早進行基因篩檢,以能及早用藥,提高治療效果。(影片截圖)
SMA患者因脊髓運動神經細胞受損,影響抬頭、坐立、行走、說話、呼吸及進食等活動功能,嚴重會導致全身肌肉無力及無法呼吸而死亡。台灣健保於2010年7月起,為SMA患者支付脊髓注射藥物的費用,再於2023年4月支付口服藥物的費用,兩者屬於維持性的治療,1年的藥費約500萬至550萬元新台幣(約121萬至133萬港元)。
及後於今年8月,當局再將一次性基因治療藥物納入支付名單,資助對象是6個月以下病發及帶有基因突變的病童,並估計1年有8至9人受惠。於台灣接受資助的首宗個案,是一名來自高雄的男嬰,他於第12日確診SMA,治療前頭部無法直立,全身軟弱無力,而且哭聲微弱,令其父母相當難過。
今年8月時,4個月11日大的男嬰正式接受相關藥物治療,治療25日後已能可以抬頭;經過2個月後,頭部已經可以控制自如,並且可以坐立、手部抓握力變強,並且可以連續翻身、自己抬頭。男嬰的父母非常感謝當局的資助,現時可以聽到兒子洪亮的哭聲,有精神地在床上翻滾;他們期望兒子可以一步一步站起來,甚至行走、跑步、上學。
高雄醫學大學附設中和紀念醫院教授鐘育志指,SMA曾經被視為無法醫治的神經退化疾病,現時則有藥物治療,不過由於一次性基因治療藥物費用高昂,因此男嬰接受注射的過程中,醫療團隊都小心翼翼地進行。台灣健保署指,為有效預防及治療SMA,新生嬰兒應及早進行基因篩檢,以能及早用藥,提高治療效果。
來源:《自由時報》
更多文章:




